文献解读
TJB2015-揭示水稻花药发育过程中调控蛋白的广泛磷酸化
发布日期:2020-06-01 5:50 浏览: 5,656 views次
花药的发育,特别是在减数分裂时期,对植物有性生殖至关重要。同时,体细胞(特别是绒毡层)与减数分裂细胞之间的细胞间通讯对体细胞花药发育和减数分裂都具有重要意义。水稻(Oryza sativa)是世界主要农作物之一,为全球一半以上的人口提供粮食,因此在人口爆炸和气候变化的情况下,保持和提高水稻产量以保障粮食安全是一个巨大的挑战。此外,水稻也是研究单子叶植物发育过程的分子和遗传机制的良好模型。为了研究在花药发育过程中调节蛋白质活性的分子机制,作者采用基于高分辨率质谱的蛋白质组学和磷酸化蛋白质组学分析方法,对水稻减数分裂前后的花药发育进行了研究。一共鉴定了4984个蛋白和3203个磷酸化蛋白,其中8973个磷酸化位点(p位点)。在本文检测到的这些蛋白中,1544个磷酸化蛋白目前在植物蛋白磷酸化数据库(P3 DB)中缺失。通过Mapman富集分析表明,磷酸化蛋白中DNA修复、转录调控和信号转导相关蛋白的表达量过高。共检测到10个经基因鉴定的水稻减数分裂蛋白在25个p-位点磷酸化,发现400多个减数分裂蛋白磷酸化,并对其磷酸化位点进行了精确定位。163种可能在细胞间通讯中起作用的假定分泌蛋白也被磷酸化。同时发现磷酸化对DNA合成、RNA剪接和RNA导向的DNA甲基化途径有着广泛的影响。本文数据支持由水稻激酶-蛋白质相互作用图谱预测的46对激酶-底物,SnRK1底物高度富集。

1.RAM蛋白质组和磷蛋白组的整体分析
作者基于含有减数分裂花药的水稻圆锥花序的形态学特征,从野生型水稻植物中一共收集了10000个RAM用于进一步的实验。然后通过蛋白质组学和磷酸化蛋白质组学的方法一共鉴定得到24087个肽,代表4984个非冗余单个蛋白质。在该研究中,总共检测到1650个基因与微阵列分析中的表达数据。
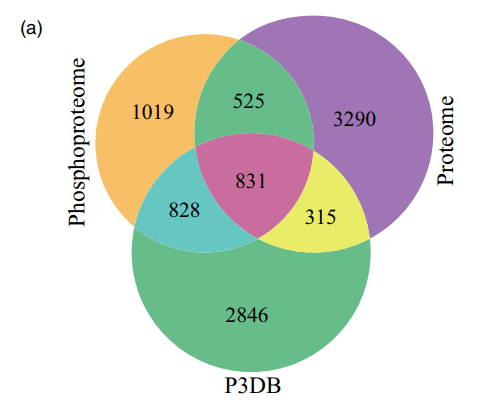
同时,通过磷酸化蛋白质组学鉴定得到3203个蛋白质和8973个磷酸化位点,其中有9594个独特的磷酸肽,1847个磷酸化蛋白仅存在于磷酸蛋白质组中。结合两个数据集,总共鉴定了RAMs中的6831个蛋白质。与植物蛋白质磷酸化数据库(P3DB)的比较显示,当前P3DB中没有本文RAMs数据集中的1544个磷酸蛋白质(图1A)。
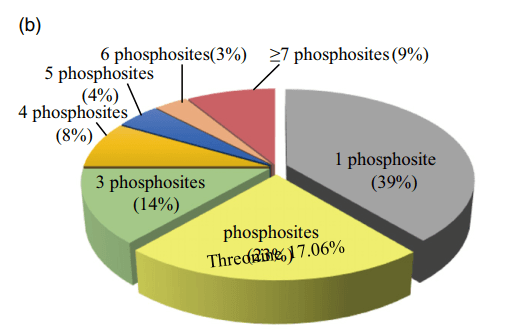
特定磷蛋白中磷酸化位点的数目变化很大。LOC_Os01g04650的磷酸化位点最多为34个,编码一个含有PB1结构域的蛋白质(图1B),39%的磷酸化位点有一个检测到的磷酸化位点,23%的磷酸化位点有两个。约9%(275个蛋白质)的已鉴定磷酸化蛋白质具有超过7个磷酸化位点,包括36个转录因子,至于磷酸化氨基酸残基,在8973个已确定的磷酸化位点中,81.33%(7236)在丝氨酸(pSer)上磷酸化,17.06%(1518)在苏氨酸(pThr)上磷酸化,1.6%(143)在酪氨酸(pTyr)上磷酸化。
2. RAM蛋白质组和磷蛋白组的细胞定位和功能预测
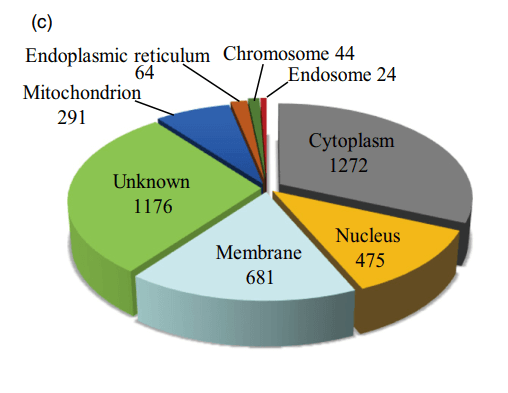
作者基于细胞成分进行了GO分析对蛋白质组和磷蛋白组鉴定的蛋白质的亚细胞定位进行生物信息学分析,蛋白质组和磷蛋白组的分布模式相似,最大的比例分配给细胞质,其次是膜和细胞核。 值得注意的是,在蛋白质组和磷蛋白组中分别发现了52和44个染色体相关蛋白(图1C)。
作者通过MapMan分析对蛋白质组和磷酸化蛋白质组进行功能分类富集预测分子功能。在RAM蛋白质组中富集了各种分子功能,包括DNA、氨基酸代谢、蛋白质、细胞、糖酵解、TCA、核苷酸代谢、氧化还原和脂质代谢。相反,在RAM的磷酸化蛋白质组中,包含RNA、DNA、细胞和信号转导的四个类别的蛋白显著地高表达。进一步分析过度表达的种类的详细信息,发现在磷酸蛋白质组和蛋白质组中有9个功能过程被富集(图1D)。
3. 减数分裂和假定减数分裂的蛋白质
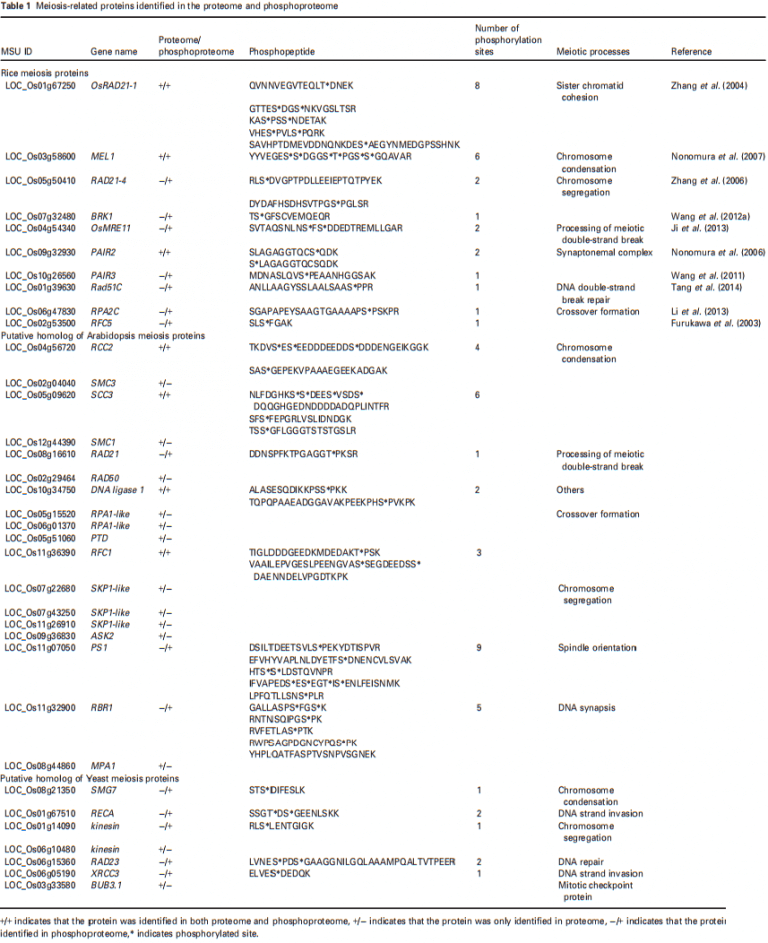
本文从磷酸蛋白质组16个非冗余磷酸肽中鉴定出23个pSer和2个pThr位点,涵盖了10个已知的减数分裂蛋白;这些蛋白质对染色体缩合(MEL1)、同源配对和突触(PAIR2和PAIR3)、DNA DSB修复(RAD51C和OsMER11)、交叉形成(RPA2C和RFC5)和染色体分离(RAD21-4和BRK1)非常重要(表1),同时发现OsMEL1在同一肽中有6个磷酸化位点(5个pSer和1个pThr)。
作者在拟南芥和酵母中寻找已知减数分裂基因的水稻同源基因。在蛋白质组和磷酸化蛋白质组中发现了14个拟南芥减数分裂蛋白的17个水稻同源物。其中3个为水稻SKP1样同源物,2个为RPA1同源物;另外7个水稻同源物,包括RCC2、SCC3、RAD21、DNA连接酶1、RFC1、PS1和RBR1被磷酸化,共鉴定出26个磷酸化位点,其中23个pSer和3个pThr位点。一种蛋白质中磷酸化位点最多的是9个,分布在PS1的5种不同的磷酸肽中。蛋白质组和磷酸化蛋白质组中鉴定了七个酵母减数分裂蛋白的水稻同源物。其中,5个被磷酸化,包括SMG7、RECA、驱动蛋(LOC_Os01g14090)、RAD23和XRCC3,有7个磷酸化位点(6个在Ser上,1个在Thr上)(表1)。
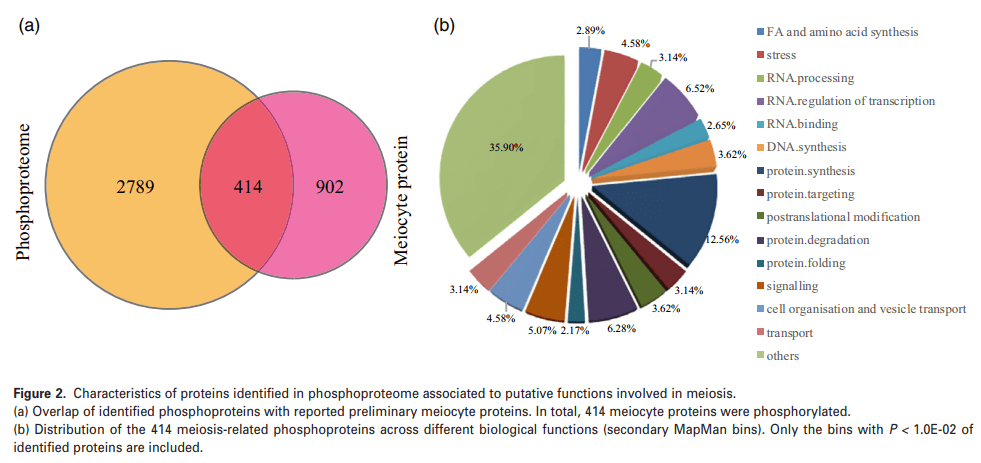
通过早稻雄性减数分裂细胞的蛋白质组分析报告(鉴定了1316个表达蛋白)与本文磷酸化蛋白质组学数据的比较分析表明,414个减数分裂细胞表达的蛋白质被磷酸化(图2A)。通过MapMan分析表明,它们主要分为DNA和蛋白质合成、RNA处理和转录调控、蛋白质降解、靶向和翻译后修饰以及信号转导(图2B)。进一步分析发现,除表1所列减数分裂相关蛋白外,另外15个磷酸化蛋白被注释为染色质相关蛋白。同时将117个磷酸化蛋白分为蛋白质合成组,包括核糖体蛋白、起始和伸长因子亚组,其中52个在水稻早期减数分裂中被鉴定。
4.DNA合成与RNA剪接途径
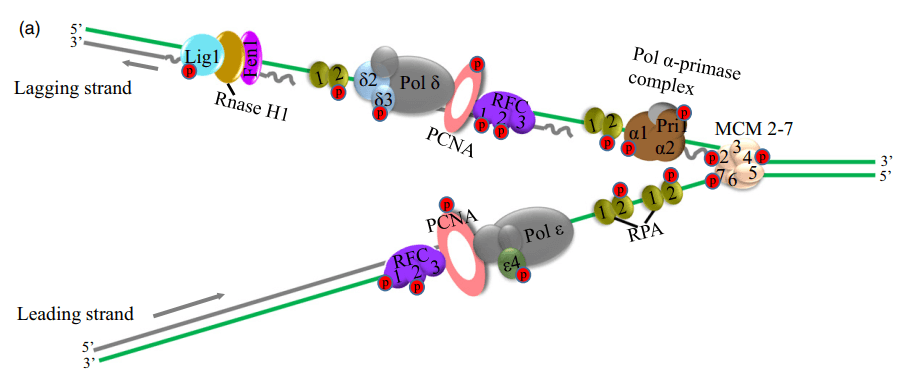
大多数与DNA合成相关的蛋白质在本文数据集中被鉴定出来,包括在DNA合成起始过程中起关键作用的小染色体维持蛋白(MCM2-7)复合物的所有亚单位(图3A),与之前的报道一致,细胞周期蛋白依赖性激酶(CDK)介导的MCMs磷酸化影响其在爪蟾卵和HeLa细胞中的染色质负载和螺旋酶激活。
作者在RFC复合物和增殖细胞核抗原(PCNA)中检测到RFC1和RFC2的磷酸化(图3A)。同时许多参与滞后链合成的效应子被发现磷酸化,例如Pol a和d的亚基。
据估计,79%的拟南芥基因至少有一个内含子;进一步分析表明,近25%的多外显子基因在开花期间经历了选择性RNA剪接,一项RNAseq分析发现,13000多个新的选择性剪接RNA在减数分裂细胞中高度表达。拟南芥剪接体由107个蛋白质组成,在该研究中检测到87个拟南芥剪接体蛋白质的水稻同源物,其中52个被磷酸化(图3B)。13个拟南芥SR蛋白的水稻同源物被磷酸化,包括SR1的2个、SRZ33的4个、SRZ21的2个、SR45.1的1个、SCI1的1个、SC35样剪接因子的1个和AT4G36980。
SR1的两个水稻同源基因LOC_Os07g47630.1和LOC_Os03g22380.1在RNA识别基序中分别在丝氨酸88和丝氨酸123上磷酸化,但在RS结构域中没有磷酸化。水稻SRPK1、OsSRPK4的同源物在激酶结构域中也被丝氨酸390磷酸化。同时SR45.1的水稻同源基因LOC_Os01g26940.1处于高磷酸化状态,含有17个pSer位点(S5、S86、S100、S193、S228、S284、S296、S301、S307、S323、S320、S355、S392、S433、S481、S583、S603)和1个pThr位点(T282),这些磷酸化位点分布于RS1、RRM和RS2结构域中。
5. 分泌蛋白
在24种具有细胞-细胞通讯功能的筋膜素样蛋白中,本文鉴定了9种蛋白质,包括MTR1。同时163种推测的分泌蛋白被磷酸化。其中2个是富含半胱氨酸的分泌蛋白(LOC_Os04g44600和LOC_Os04g58120),分别是拟南芥表皮模式因子(EPFL)1和EPF2的同源物,在6个和5个位点磷酸化。EPFL1和EPF2作为受体样蛋白激酶ER、ERL1和ERL2的配体,在拟南芥TPD1同源物OsTDL1B(LOC_Os10g14020)的N端发现了两个磷酸化位点,这些磷酸化位点在OsTDL1A和TPD1之间不保守。
6. 蛋白激酶和磷酸酶
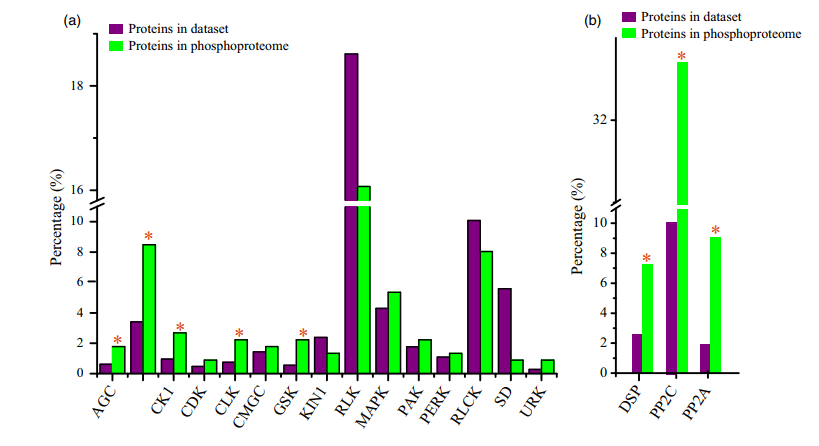
作者发现蛋白质激酶在磷酸化蛋白质组中的表达量非常高(7.0%),而在整个预测的蛋白质组中的表达量为2.2%。在磷酸蛋白质组数据集中,共鉴定出224个激酶和539个磷酸化位点,占水稻蛋白质数据库中1467个总注释激酶的15%。通过对不同的激酶家族进行富集分析确定控制磷酸化调控网络的激酶家族,受体样激酶(RLKs)家族所占比例最大(74,33%),其次是CAMK(19,8.5%)和MAPK(12,5.3%)(图4A)。同时GSK、URK、CLK、AGC、CK1和CAMK家族中的磷酸化蛋白所占比例高于在其他家族中。一共检测到55种磷酸化磷酸酶,共有121个磷酸化位点(图4B),其中PP2C磷酸酶所占比例最大(18,32.7%),其次是PP2A(8,14.5%)、DSP(6,10.9%)和其他磷酸酶。
9个Rlk(受体样激酶),包括EMS1、SERK1、SERK2、BAM1、BAM2、ER、ERL1、ERL2和RPK2,已被揭示在拟南芥花药发育中起关键作用。在水稻花药磷酸化蛋白组中,共鉴定出12个亚科74个Rlk,161个磷酸化位点。同时在36个磷酸化LRR-rlk中,15个属于LRR-III,5个被划分为LRR-V亚家族。在55种已鉴定的磷酸酶中,有11种具有3个以上的磷酸化位点,其中3种在9个位点磷酸化。这三种磷酸酶的序列比对表明它们具有70%的序列同源性。此外,8个磷酸化位点高度保守。
作者采用水稻激酶-蛋白质相互作用图提出的激酶-底物关系去了解蛋白质磷酸化介导的调控网络。在磷酸化蛋白质组中发现了46对激酶-底物,并用CYTOSCAPE 2.8软件说明了相互作用网络,属于SNF1相关蛋白激酶-1(SnRK1)家族的两个亚单位SnRK1a/OSK1和SnRK1b/OSK3的底物最为丰富。此外,还发现大麦SnRK1a和SnRK1b在花药中表达,反义SnRK1的表达导致大麦花粉发育异常和雄性不育。该研究中上游磷酸酶PP2Cc和SnRK1复合物的其他三个亚单位,KINI,oscbs8/KINbc和一个DUF581结构域(LOC_Os08g31510)被鉴定。另外,两种转录因子(一种MYB样转录因子(LOC_Os02g57200)和一种三螺旋转录因子(LOC_Os02g33770)以及三种功能未知的蛋白质被认为是SnRK1的底物,SnRK1可能通过磷酸化下游底物在调控RAM发育中发挥重要作用。同时发现了三种mapk(MAPK12-2、MAPK14和MAPK20-4)的10种底物以及MAPK6和MKK6的上游激酶(图4C)。
7. 转录因子(TFs)
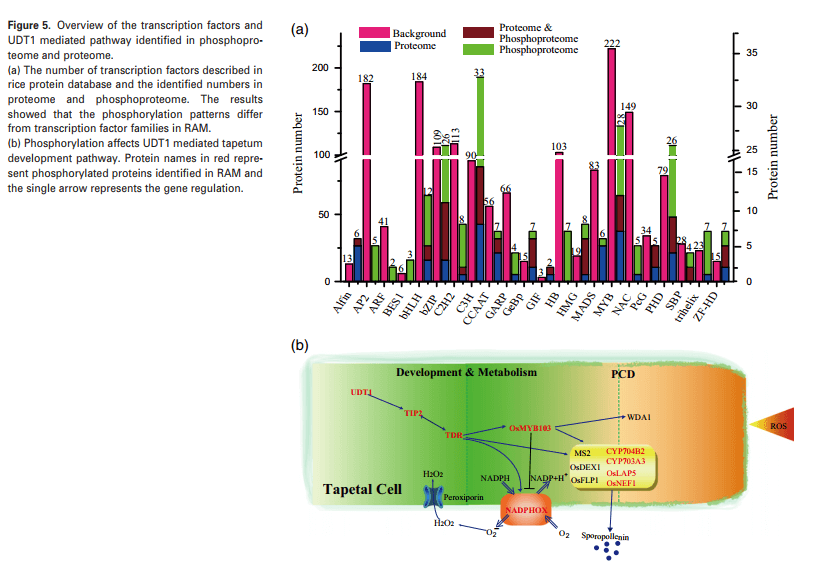
作者通过本文发现230个TFs在RAM中磷酸化(图5A)。这些磷酸化TFs被分为不同的家族,包括C3H(25,10%)、bZIP(23,10%)、PHD(22,9.5%)、MYB(21,9%)和bHLH(9,4%)。同时BES1、HMG、GIF、GeBp、ZF-HD、SBP、C3H和bZIP亚家族的磷酸化比率高于其他亚家族。对于23个磷酸化bZIP TFs,检测到88个磷酸化位点。同时发现更多的成员在IX(8个1)和VI(7个1)亚家族中被磷酸化。同时,在24个磷酸化的C3H-TFs中,V亚家族的所有三个成员(OsC3H22、OsC3H23和OsC3H53)被磷酸化。
在这项研究中,蛋白质组学和磷酸化蛋白质组学数据已经鉴定出19种已知参与水稻花药发育的蛋白质;其中9种蛋白质在16个位点磷酸化。
TFs、UDT1、TDR和OsMYB103的水稻同源物在6个位点被发现磷酸化(图5B)。在拟南芥中,绒毡层特异性NADPH氧化酶(RBOHE)最近被报道对绒毡层程序性细胞死亡(PCD)至关重要。在本文数据集中水稻NADPH氧化酶(OsRBOHE,LOC_Os08g35210)被磷酸化。
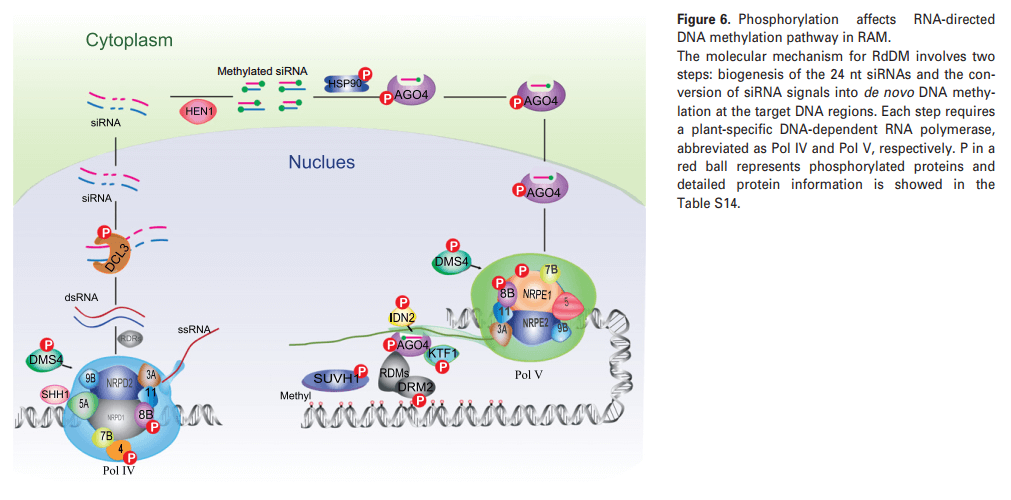
8. RNA定向DNA甲基化(RdDM)途径
作者确定了RAM中RdDM途径的大多数因素(图6),在磷酸化蛋白质组学数据中鉴定了在155个位点磷酸化的37个因子,包括域重排甲基酶2(OsDRM2)、类Dicer 3(DCL3)、Argonaute 4(AGO4)和NRPD 4。作者一共鉴定了13个AGO蛋白,其中9个被磷酸化,15个Snf2蛋白中13个被磷酸化。同时检测到其他三种染色质重塑因子的磷酸化作用,它们是KOW结构域转录因子1(KTF1)的水稻同源物。在水稻IDN2同源物(LOC_Os01g44230)的N端XS结构域中检测到3个磷酸化位点(Ser 5、Ser 9和Ser 11)。
玉米生殖细胞(减数分裂前)和体细胞的比较转录分析表明,RdDM途径中的基因,包括AGO4和IDN2,优先富集在生殖细胞中。