
技术文档
蛋白质组学中蛋白质和肽段分离方法的比较
 设计基于蛋白质组学的串联质谱实验需要选择最好的蛋白质和肽段的分离方式这有利于得到最佳的蛋白质覆盖率。但是一个研究者如何确定哪个是最佳的方法呢?Mostovenko et al. (2013)提供了一个灵活方法探索这个难题。全面理解这三个最主流的方法-阳离子交换色谱(SCX),肽等电点聚焦(pIEF)和聚丙烯酰胺凝胶电泳(sds – page)得出最好的结论,以及这些方法最优的应用条件。
设计基于蛋白质组学的串联质谱实验需要选择最好的蛋白质和肽段的分离方式这有利于得到最佳的蛋白质覆盖率。但是一个研究者如何确定哪个是最佳的方法呢?Mostovenko et al. (2013)提供了一个灵活方法探索这个难题。全面理解这三个最主流的方法-阳离子交换色谱(SCX),肽等电点聚焦(pIEF)和聚丙烯酰胺凝胶电泳(sds – page)得出最好的结论,以及这些方法最优的应用条件。
为了一致性,除了比较每一种方法时使用标准的样品,这些研究者也关注每一个过程的有效附加信息。因为初步的分级常用作减少MS分析的初始样品的复杂程度。每一个碎片的物理特性可以通过不同的方法获得,这些信息常用于改善源于原始光谱文件的肽段匹配和预测。例如,SDS-PAGE根据分子量分离,从而为MS做准备,然而pIEF是等电点分离。
为了实验的一致性,科学家使用两种样品-大肠杆菌细胞裂解液和人体血浆做分析。每个样品经过适当的酶解,(例如经过SDS-PAGE分离之后是胶内酶解,之后是pIEF和SCX分级)SDS-PAGE和pIEF都有在标准操作流程。SCX的分级是使用Dionex UltiMate 3000 LC 系统 (Thermo Scientific)。Mostovenko和他的同事,接着用超高效液相色谱(PepMap C18 columns, Thermo Scientific)串联质谱分析检验肽段的酶解情况。团队将所有数据用Taverna workflow整合到一起。
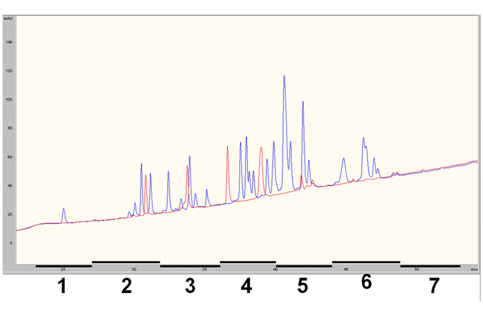
这个结果显示,SCX分级方法能得到最好的蛋白组覆盖率。用这种方法处理大肠杆菌样品,从6731个鉴定到的肽段匹配到1139个蛋白,在人体血浆样品中可从1642个肽段中匹配到183个蛋白。将这个结果与UniProt FASTA整个测序数据库作比较,显示两种样品各自蛋白覆盖率为35%和29%.使用SDS-PAGE方法的结果则是34%和23%,pIEF则是27%和24%。
研究者接着检验,从不同方法得到的额外信息是否能够改善数据结果。他们发现,通过调节PH,根据这些参数分离这些碎片呈现一致的线性,他们可以确定在pIEF分级方法中的出现的异常值。同样的,通过原蛋白分子量与预测鉴定的肽段和凝胶切片位点作比较也是有助于SDS-PAGE预备样品的异常值检测。
尽管实验结果明显显示SCX是最好的蛋白组特性分级方法,但研究人员认为原始的样品载入可能是这个方法的一个限制性因素。例如,SDS-PAGE的最佳上样量是30µg,然而对于pIEF是50µg,在SCX中较多达到100µg。他们承认即便保持LC-MS/MS的标准参数恒定和系统的稳定性,也不可能使每种分级方法达到最佳的操作条件,因此应相互协调。
作者认为:尽管实验条件的限制,用Taverna综合的分析是成功的,允许比较这三个独立的数据库是有效的。更重要的是,当决定使用哪种方法时,他们发现考虑每个分级过程额外信息的价值是非常重要的,因为每个考虑都能改变采集到的蛋白组学数据。
Reference
- Mostovenko, E., et al. (2013) “Comparison of peptide and protein fractionation methods in proteomics“, EuPA Open Proteomics, 1 (pp. 30–7), doi: 10.1016/j.euprot.2013.09.001.